
Amyloidosis is a rare disease. Most people have never heard of it until they or their family member of friend is diagnosed. Many doctors know little about the condition. Patients, friends, family and carers often have many questions. On this frequently asked questions page we have tried to collect all the most frequently asked questions and concerns in one place. We hope that having this information accessible will help to inform and allay worry as much as possible.
The first section addresses very common concerns affecting all patients with amyloidosis, such as life expectancy with amyloidosis , the logistics of National Amyloidosis Centre appointments, and what to expect at your appointment. The other sections are divided according to the different types of amyloidosis- AL amyloidosis, ATTR amyloidosis, AA amyloidosis, AFib amyloidosis, localised amyloidosis and ALECT2 amyloidosis. These are very different diseases, with different treatments. To avoid unnecessary worry, we recommend trying to avoid reading too much until you know for sure which type of amyloidosis you or your family member has.
To connect with other patients with amyloidosis and their families and carers, visit the NAC patient support forum. The forum has been running since 2015 and now numbers over 1200 members. It has enabled patients, family members and carers from all over the UK nd abroad to meet and give each other support, advice and encouragement in cyberspace. There are also several small local support groups organised by patients from various parts of the country. The meetings are very informal - just a chance for patients, families and carers to meet over a coffee, get to know each other and chat.
When Covid restrictions were announced in early 2019, the Scottish support group, run by Mark McConway, switched from meeting about 4 times a year for lunch to meeting weekly on Zoom throughout lockdown. They have been joined by people from the rest of the UK, Ireland, Denmark, the USA, Canada and France. For more information, contact Mark at mcconway.mark@gmail.com
Amyloidosis

- AL amyloidosis
- Wild-type ATTR amyloidosis
- Localised amyloidosis
Patient Concerns
The National Amyloidosis Centre
UK patients
Patients in the UK require a physician’s referral to the NAC. Information for referring physicians is available here. For questions regarding appointments contact: Mr Rizwan Shaukat, tel- 02074332732 rizwan.shaukat@nhs.netOverseas patients
The Royal Free Hospital and the National Amyloidosis Centre welcome overseas patients. European Union residents may be entitled to an NHS assessment in the UK under EU reciprocal arrangements for medical care that is not available locally (EU S2 form). Non‑NHS entitled patients are welcome but are usually liable to charges. Information for referring physicians is available here: . For inquiries regarding NHS entitlement or charges, contact: Mr Rizwan Shaukat, tel- 02074332732 rizwan.shaukat@nhs.net- blood and urine tests
- ECG and echocardiogram (ultrasound scan of the heart)
- whole body SAP scan to establish the distribution and quantity of amyloid deposits
- additional tests in some patients may include:
- abdominal fat biopsy
- bone marrow biopsy
- DPD scan of the heart
- cardiac MRI
- physician evaluation
- specialist nurse consultation
Symptoms
AL amyloidosis symptoms
In AL amyloidosis, amyloid deposits may affect any part of the body except for the brain. Usually one or two organs are predominantly affected (known as the “dominant” organs). Patients with AL amyloidosis may complain of general problems such as weight loss, fatigue, weakness, loss of appetite and easy bruising. They may also develop symptoms of disease affecting the kidneys, heart, nervous system, gut, liver, spleen, skin and joints. Macroglossia (enlarged tongue), bruising of the skin round the eyes (‘racoon eyes’ or ‘panda eyes’) and the shoulder pad sign (swelling of both shoulders) are quite rare, occurring in less than 15% of cases. But when these signs do occur, they are very strongly suggestive of AL amyloidosis.ATTR amyloidosis symptoms
ATTR amyloidosis mainly affects the heart and the nerves. Symptoms of heart disease may include shortness of breath, palpitations, leg swelling, weight loss, nausea, fatigue, fainting and chest pain. Symptoms of nervous system disease may include weakness, pain and loss of sensation in the arms and legs, disturbances of bowel, bladder, blood pressure and sexual function.AA amyloidosis symptoms
AA amyloidosis mainly affects the kidneys and spleen. Protein in the urine is usually the first sign. After that, more serious kidney disease can develop, including nephrotic syndrome, when very large amounts of protein in the urine make it appear frothy, and there is ankle swelling and weight gain. The spleen is often enlarged.Non-ATTR hereditary amyloidosis
Hereditary fibrinogen A alpha chain amyloidosis (Afib)
AFib amyloidosis (fibrinogen Aα-Chain amyloidosis) is a rare inherited genetic condition. People with this condition develop kidney disease caused by build-up of abnormal protein deposits called amyloid in their kidneys. Abnormal kidney function is usually first diagnosed in this condition in middle age. Kidney function then tends to become progressively worse over several years until the kidneys stop functioning altogether. ‘Renal replacement’ treatment (either dialysis or kidney transplantation) is then required. AFib amyloidosis is equally common in men and in women. In the UK it is most common in people of British Caucasian ancestry but it may occur in people of any ethnic origin. For more information see here.Hereditary apolipoprotein A1 amyloidosis (ApoA1)
ApoA1 amyloidosis usually causes high blood pressure and kidney disease. It often also affects the liver and sometimes the heart.Hereditary gelsolin amyloidosis (AGel)
AGel amyloidosis, also known as Familial Amyloidosis Finnish (FAF) type because most patients are from Finland, causes eye, skin and cranial nerve symptoms. Despite kidney amyloid deposits, kidney function is usually not affected.Hereditary lysozyme amyloidosis (ALys)
ALys amyloidosis mainly causes kidney disease, and there may also be amyloid in the stomach lining. This form of amyloid tends to build up very slowly indeed and patients can remain stable for many years.- peripheral neuropathy: limb weakness and pain, loss of sensation
- autonomic neuropathy: disturbances of bowel, bladder and blood pressure and sexual dysfunction
- heart failure – symptoms result from stiffening of the heart due to amyloid deposits (restrictive cardiomyopathy). They may include:
- shortness of breath, sometimes just after mild exertion
- palpitations and abnormal heart rhythms, most frequently atrial fibrillation or atrial flutter
- leg swelling (oedema)
- weight loss
- nausea
- fatigue
- dizziness and collapse (syncope or fainting), which may occur after exertion, or after eating
- disrupted sleep
- angina (chest pain)
- disease due to amyloid deposits in the :
- eye
- kidneys
- thyroid gland
- adrenal glands
- blood vessels
- age of onset
- rate of disease progression
- involvement of different body systems
AL Amyloidosis

Macroglossia--enlarged tongue, sometimes with bite marks
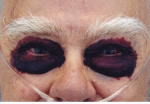
Raccoon eyes
- Reducing the supply of amyloid forming precursor proteins.
- Supporting the function of organs containing amyloid.
- age
- quantity of amyloid
- organs affected- heart and kidney function are especially important for treatment decisions
- other diseases and general health
- personal preference
- achieving a good response to treatment as rapidly as possible in order to halt the damage caused to organs by the amyloid deposits,
- minimising adverse side effects of the drugs.
- Accumulation of new amyloid ceases
- Existing amyloid deposits often regress (become smaller)
- Organ function is often preserved and may also recover
 yloid deposits may regress.
It may be helpful to envisage amyloid deposits blocking up the organs as somewhat similar to the everyday situation of a blocked sink. Water running from the tap at full blast represents the amyloidogenic free light chains pouring out of abnormal plasma cells. The small sink outlet represents the body’s limited capacity to clear amyloid deposits away from the organs. If the tap is turned on fully so that the rate of running water far exceeds the drainage rate, water builds up in the basin despite some drainage. If the tap is turned down sufficiently, the water can drain away slowly.
If the rate of light chain production can be turned down sufficiently, amyloid stops accumulating in the tissues. Amyloid deposits may even be cleared away faster than they are produced.
yloid deposits may regress.
It may be helpful to envisage amyloid deposits blocking up the organs as somewhat similar to the everyday situation of a blocked sink. Water running from the tap at full blast represents the amyloidogenic free light chains pouring out of abnormal plasma cells. The small sink outlet represents the body’s limited capacity to clear amyloid deposits away from the organs. If the tap is turned on fully so that the rate of running water far exceeds the drainage rate, water builds up in the basin despite some drainage. If the tap is turned down sufficiently, the water can drain away slowly.
If the rate of light chain production can be turned down sufficiently, amyloid stops accumulating in the tissues. Amyloid deposits may even be cleared away faster than they are produced.  In AL amyloidosis, abnormal plasma cells produce a large quantity of one particular type of light chain.
The abnormal light chains are called ‘free’ light chains because they are not linked to heavy chains within antibodies, like normal light chains. Instead, they exist freely in the bloodstream until they deposit in the organs as amyloid fibrils.
Under normal circumstances, light chain concentrations are very low in the bloodstream. Sensitive blood tests can detect abnormal free light chain concentrations in about 95% of patients with AL amyloidosis.
FLC concentration is measured to assess plasma cell response to treatment. Introduction of this test about 10 years ago was a landmark advance in the management of patients with AL amyloidosis. FLCs can be detected and measured in over 95% of AL amyloidosis patients. Blood concentration of FLCs is a sensitive measure of treatment effects on the abnormal plasma cells.
When treatment is successful, the earliest test which shows improvement is the FLC concentration.
FLC concentration drops before there is measurable improvement in the function of affected organs, before patients start to feel better and before there is any visible change in amyloid deposits seen on SAP scans.
In AL amyloidosis, abnormal plasma cells produce a large quantity of one particular type of light chain.
The abnormal light chains are called ‘free’ light chains because they are not linked to heavy chains within antibodies, like normal light chains. Instead, they exist freely in the bloodstream until they deposit in the organs as amyloid fibrils.
Under normal circumstances, light chain concentrations are very low in the bloodstream. Sensitive blood tests can detect abnormal free light chain concentrations in about 95% of patients with AL amyloidosis.
FLC concentration is measured to assess plasma cell response to treatment. Introduction of this test about 10 years ago was a landmark advance in the management of patients with AL amyloidosis. FLCs can be detected and measured in over 95% of AL amyloidosis patients. Blood concentration of FLCs is a sensitive measure of treatment effects on the abnormal plasma cells.
When treatment is successful, the earliest test which shows improvement is the FLC concentration.
FLC concentration drops before there is measurable improvement in the function of affected organs, before patients start to feel better and before there is any visible change in amyloid deposits seen on SAP scans. - If the abnormal plasma cells are secreting lambda chains, the lambda/kappa ratio is checked.
- If the abnormal plasma cells are secreting kappa chains, the kappa/ lambda ratio is checked.
- lambda (λ) chains
- kappa (κ) chains
- If a patient with AL amyloidosis has amyloid due to abnormal production of kappa chains, dFLC concentration is kappa chain concentration minus lambda chain concentration.
- If a patient with AL amyloidosis has amyloid due to abnormal production of lambda chains, dFLC concentration is lambda chain concentration minus kappa chain concentration.
- Complete response (CR): No FLCs detected (ie negative serum and urine immunofixation tests and normal FLC ratio)
- Very good partial response (VGPR): dFLC concentration below 40 mg/dl
- Partial response (PR): dFLC decrease more than 50% from previous value
Living with AL Amyloidosis
- Diet
- Diuretics
- Daily weights
Peripheral neuropathy:
Medications that may help to alleviate neuropathic pain include gabapentin, pregabalin and duloxetine. Medical staff can give advice regarding appropriate foot care and footwear. This is important in order to prevent painless ulcers at pressure points and to protect areas of the foot that lack sensation.
Autonomic neuropathy:
If there is orthostatic hypotension (decrease in blood pressure and faintness on standing up from sitting or lying positions), thigh high elastic stockings may be recommended. Drug treatment with midodrine may also be helpful. Care should be taken to avoid dehydration if there is vomiting and diarrhoea. Intravenous fluids and anti‑nausea drugs may be necessary, but it is important to avoid fluid overload if there is heart disease. There are drugs that can help to control diarrhoea and constipation, and others that can help to combat erectile dysfunction.
Some tips from a patient with neuropathy:
Dealing with postural hypotension:
1. After sitting down for more than 1/2 hour always stand for about 30 seconds before moving off.
2. If feeling light headed or you have ringing in your ears when walking, stand still for a few moments until it passes. Sometimes you can walk through it after some practice!
3. If it gets really bad always ask for help or sit down. I find it, in some cases good to hold onto my wife’s arm when walking distances.
4. I find that when I get a cold or virus the postural hypotension can get worse. I find it helps by taking the standard doses of paracetamol.
5. When getting up from bed in the morning I always sit on the side for a few moments before walking off.
6. Never run and always pace yourself.
7. Always allow more time than you think you need
Dealing with peripheral neuropathy symptoms:
1. I find doing weekly muscle build-up and light exercise at the gym helps a lot.
2. I find having a regular leg and feet massage eases my leg pains and stiffness.
3. Keep your legs and feet always moisturised.
4. Carry out daily hand exercise to keep them working.
Lifestyle Advice for Patients with AL Amyloidosis
- frequent handwashing, especially before handling food
- wash all surfaces, cutting boards and cutting utensils thoroughly
- Keep hot food hot and cold food cold.
 These instructions are not comprehensive and you should always ask your health care providers for personalised advice and recommendations.
These instructions are not comprehensive and you should always ask your health care providers for personalised advice and recommendations. ATTR Amyloidosis
Hereditary ATTR Amyloidosis
- Peripheral neuropathy: limb weakness and pain, loss of sensation.
- Autonomic neuropathy: disturbances of bowel, bladder and blood pressure and sexual dysfunction.
- Heart failure - symptoms result from stiffening of the heart due to amyloid deposits (restrictive cardiomyopathy). They may include:
- shortness of breath, sometimes just after mild exertion
- palpitations and abnormal heart rhythms, most frequently atrial fibrillation or atrial flutter
- ankle swelling (oedema)
- fatigue
- dizziness and collapse (syncope or fainting), which may occur after exertion, or after eating
- angina (chest pain)
- weight loss
- nausea
- disrupted sleep
- disease due to amyloid deposits in the :
- eye
- kidneys
- thyroid gland
- adrenal glands
- blood vessels
- age of onset
- rate of disease progression
- involvement of different body systems
Treatment of hereditary ATTR amyloidosis is discussed in the section below.
Here are some tips for dealing with common symptoms, from a patient who has suffered from this condition for many years:
Dealing with postural hypotension
1. After sitting down for more than 1/2 hour always stand for about 30 seconds before moving off.
2. If feeling light headed or you have ringing in your ears when walking, stand still for a few moments until it passes. Sometimes you can walk through it after some practice!
3. If it gets really bad always ask for help or sit down. I find it, in some cases good to hold onto my wife’s arm when walking distances.
4. I find that when I get a cold or virus the postural hypotension can get worse. I find it helps by taking the standard doses of paracetamol.
5. When getting up from bed in the morning I always sit on the side for a few moments before walking off.
6. Never run and always pace yourself.
7. Always allow more time than you think you need!
Dealing with painful neuropathy symptoms
1. I find doing weekly muscle build-up and light exercise at the gym helps a lot.
2. I find having a regular leg and feet massage eases my leg pains and stiffness.
3. Keep your legs and feet always moisturised.
4. Carry out daily hand exercise to keep them working.
Diet
Over the last 6 years since having my liver transplant I have found that by adjusting my diet my bowel problems have reduced:
1. By reducing the amount of wheat has helped a lot. I very rarely eat sandwiches, cakes and eat very little pastry.
2. I try not to have processed foods and have cooked food at lunchtime and in the evenings.
3. I have cut out sugar as much as I can. (Chocolate seems to still be good!!!)
4. I have cut out all beer and lager and tend to drink water, wine and some spirits.
5. I restrict to only having mild curries.
Wild-type ATTR Amyloidosis
- shortness of breath, sometimes just after mild exertion
- palpitations and abnormal heart rhythms, most frequently atrial fibrillation/ flutter
- ankle swelling (oedema)
- fatigue
- dizziness or fainting, which may occur after exertion, or after eating
- angina (chest pain)
- weight loss
- nausea
- disrupted sleep
- Tissue biopsy
- Genetic testing
- Imaging studies
Treatment of ATTR Amyloidosis
- Reducing the supply of amyloid forming precursor proteins.
- Supporting the function of organs containing amyloid
- existing amyloid deposits often regress (become smaller)
- new amyloid deposits stop appearing
- organ function is often preserved and may also recover
Reducing variant TTR supply
Genetic-based therapies
Tafamidis
Tafamidis was developed as a specific drug for ATTR amyloidosis. It is bound by TTR in the blood. This binding is thought to stabilise the TTR and makes it less amyloidogenic. The pivotal trial of tafamidis included 441 patients, some of whom had wild-type ATTR amyloidosis while others had hereditary ATTR amyloidosis. Patients who received the active drug had better outcomes than those who received placebo, including fewer hospitalisations for heart disease, a 30% reduction in death over a period of 2.5 years, reduced decline in functional capacity and improved quality of life. Tafamidis is approved in Europe for treatment of hereditary ATTR amyloidosis patients with stage 1 symptomatic polyneuropathy to delay neurological impairment and before liver transplantation, but it is not currently available within the NHS. The US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have approved tafamidis for cardiomyopathy caused by ATTR amyloidosis although it is not approved in the US for ATTR polyneuropathy. It will need to be evaluated by NICE before it can become available within the NHS.Diflunisal
This belongs to a class of drugs called ‘non-steroidal anti-inflammatory drugs’ (NSAIDs). These drugs are in common use as pain killers, for conditions such as arthritis. Diflunisal is bound by TTR in the blood. This binding is presumed to make the TTR less amyloidogenic. Trials are currently underway to assess the effect of diflunisal on the progression of neuropathy and cardiomyopathy in patients with hereditary ATTR amyloidosis. Results from the first study report were encouraging, but the numbers of patients involved was small and the extent of benefit was modest. The trial involved 130 patients with hereditary ATTR amyloidosis affecting the nerves, 64 of whom received diflunisal for two years while 66 received placebo (dummy pills). The rate of progression of neuropathy was slower in the patients who received diflunisal than in those who did not. Results of trials of diflunisal in cardiac ATTR amyloidosis are not yet available. It is important to note that NSAIDs such as diflunisal may have serious side effects, which may be especially dangerous in patients who are already unwell with amyloidosis. These side effects include: bleeding from the stomach and gut. worsening of kidney function. worsening of heart failure. Diflunisal use for ATTR amyloidosis is an ‘off-label’ indication, and only amyloidosis specialists should prescribe it. The NAC can now offer patients with ATTR amyloidosis, subject to various eligibility criteria demanded by the pharmaceutical companies, various opportunities for treatment with the new drugs or for participation in clinical trials. for information on ongoing trials at the NAC, see here.Liver transplantation
All the TTR in the blood, which forms the amyloid deposits everywhere except in the eye and the blood vessels around the brain, is made in the liver. In the past, liver transplantation was a treatment option for some patients with hereditary ATTR amyloidosis, although almost exclusively younger patients with the Val30Met mutation. Since the advent of the new drugs, liver transplantation is rarely recommended in the UK.Supporting amyloidotic organ function
In all types of amyloidosis it is important that treatment should support the function of organs containing amyloid. In ATTR amyloidosis this may include:Treatment for heart disease
ATTR amyloid deposits in the heart cause the heart to stiffen which can lead to symptoms of heart failure. Patients can benefit from supportive treatment measures for heart failure. However many standard medications used for heart failure are not helpful for patients with cardiac amyloidosis. Careful attention to fluid balance is important, as explained in the question below. In patients with low blood pressure, drugs such as fludrocortisone or midodrine may help to maintain blood pressure and allow higher diuretic doses. Some patients may experience light-headedness, fatigue on minimal exertion or fainting due to drops in blood pressure. They may benefit from instruction in how to change position carefully from lying to sitting, sitting to standing and standing to walking.Heart transplantation
For hereditary, 'variant' ATTR amyloidosis, combined heart and liver transplant has been performed in a few dozen cases around the world. This operation is only an option for a minority of patients, and it carries significant risks. Most patients with wild-type ATTR amyloidosis are too elderly to undergo a heart transplant. The risk of complications from this major operation is high with advanced age. But heart transplantation may be an option for younger, otherwise healthy patients with this condition. The 2 patients who presented to the NAC before age 60 with wild type ATTR amyloidosis survived 10 and 20 years after heart transplantation.Treatment of peripheral neuropathy symptoms
Medications that may help to alleviate neuropathic pain include gabapentin, pregabalin and duloxetine. Medical staff can give advice regarding appropriate foot care and footwear. This is important in order to prevent painless ulcers at pressure points and to protect areas of the foot that lack sensation.Treatment of autonomic neuropathy symptoms
If there is orthostatic hypotension (drops in blood pressure and faintness on standing up from sitting or lying positions), elastic stockings may be recommended. Drug treatment with midodrine may also be helpful. Care should be taken to avoid dehydration if there is vomiting and diarrhoea. Intravenous fluids and anti‑nausea drugs may be necessary, but it is important to avoid fluid overload if there is heart disease. There are drugs that can help to control diarrhoea and constipation, and others that can help to combat erectile dysfunction. The place of new drugs in the treatment of ATTR amyloidosis is discussed separately in other questions in this section. Here are some tips for dealing with common symptoms encountered in hereditary ATTR amyloidosis, from a patient who has suffered from this condition for many years: Dealing with postural hypotensionMany patients with ATTR cardiac amyloidosis should limit their fluid intake. This advice is extremely important, but is often overlooked.
The most important principle of treatment for cardiac amyloidosis is strict fluid balance control. Specialist heart failure nurse involvement may help patients to achieve this.
When there is cardiac amyloidosis, the heart may be too stiff to pump the blood efficiently around the body. This can lead to fluid build- up, causing leg swelling (oedema) and breathlessness due to fluid in the lungs. This problem is exacerbated if the patient drinks too much fluid.
Fluid excess can be avoided by careful attention to the 3 Ds:
- Diet
- Diuretics
- Daily weights
- Diet:
Fluid intake should be steady and should usually not exceed 1.5 litres per day.
Salt intake should be limited. This includes attention not just to salt deliberately added to the food during cooking or at the table but also to ready prepared foods with high salt content such as processed foods, crisps, bacon, canned meats, sausages, canned soups and smoked fish. Apart from that, a balanced, healthy diet is always advisable. It can be very helpful to meet with a dietician for precise and personalised dietary advice.
- Diuretics:
Doctors will often prescribe diuretics (water tablets) which increase the amount of urine produced and help the body to lose excess salt and water in the urine. This can help to reduce ankle swelling and breathlessness. Diuretics prescribed may include furosemide and spironolactone. Taking these drugs is not a substitute for avoidance of excessive dietary salt and water.
Patients should follow their doctor’s advice carefully regarding the dose of diuretic and the time of day when the tablet should be taken.
- Daily weights:
Some patients benefit from recording their weight regularly, usually daily or weekly. It is important that weight should be measured consistently - using the same scales, at the same time of day. This is usually best done first thing in the morning after passing urine, just wearing underclothes. Several litres of fluid can accumulate in the body without it being very noticeable. An increase in weight can be an early sign of fluid overload. The doctor or nurse can then recommend appropriate measures such as increased diuretic dose, before the patient even feels unwell because of the fluid overload.